Principles of Protein Structures
- Group research: Design of protein molecules for novel functional and behavioural activities. Also study proteins using variety of techniques like ML and computational design.
Moplecular Interactions in Biomolecules
- Non-covalent interactions modelled via Lennard-Jones, consists of van der Waals interactions, hydrogen bonds.
- Two neutral atoms in close proximity get polarised !
- Van der waals radius is r1 + r2 and is the point where the repulsive interactions become dominent.
- Hydrogen bonds can be thought of as a dipole-dipole interaction but on a molecular scale.
- Iconic interactions are the strongest, however, water ions severly reduce electrostatic interaction.
- In vacuum its ~50 kJ/mol, whereas in water its ~6 kJ/mol.
Moleculaes of Life
- Nucleic Acids
- Proteins : Made of Peptides
- Lipids : Fats
- Glycans: Sugars
Proteins have an amino group, a carboxyl group, and 20 different kinds of R groups.
They are built out of 20 amino acids encoded in DNA.
The amino acids can be classified based on the type of Carboxyl group.
- Polar and non-polar amino acids. Polar amino acids can form hydrogen bonds with itself and other amino acids. They are also hydrophobic.
- Negatively and positively charged amino acids. Their sidechains are charged at pH=7.
Synthesis of protein: condensation reaction where amino acid of one …TODO
The sequence of a protein is written from the N-terminus (one of the free $NH_3$) to the C-terminius (the one with $COO^-$).
backbobne + sidechain + actual witten structure + carboxyl extension.
Hydrophobicity of amino acids is an important feature.
Residue: the central kernal of the proteins. Find out why a Residue is called a residue. TODO
Some amino acids are special:
Cysteine can form disulphide bridges.
flowchart LR Cysteine -."+".- Cysteine2(Cysteine) --Oxidation--> Cystine Cystine --Reduction--> Cysteine2Glycine doesn’t have a sidechain group.
Proline’s sidechain group is covalently bound to the nitrogen of the peptide bond.
Protein Secondary Structures
$\alpha$-helix form to stabilize the hydrophobic core. It forms by making H-bonds between Hydrogen and oxygen.
$\beta$-sheets form hydrogen bonds with its whole backbone.
Usually the alpha and beta sheets are sometimes misaligned and imperfect.
Alpha-helix
- In the alpha-helix, the carbonyl oxygen of residue “i” forms a hydrogen bond with the amide of the residue “i+4”.
- There are only right handed alpha-helixes, except for a handful of exceptions.
Beta-sheets
- Can be wither parallel and antiparallel. They have roughly the same binding energy.
- In a beta-sheet, carbonyl oxygen and amides form hydrogen bonds between the strands, i.e between amino acids far away from each other in primary sequence.
Proteins can be composed of one of the two secondary structures or be mixed.
Beyond Secondary Structures
- The tertiary structure, which is the three-dimensional organisation of the secondary structure elements. It is also referred to as the protein fold.
- Quaternary structures refers to the association of different polypeptide chains (subunits) into a multinumeric complex.
- Protein domains are fundamental units of tertiary structue. it forms an independnt structural domin. They are often units of function.
- Domains have the same fold yet different amino acid sequences.
- Protein domains are very stable to mutations. ANd they have been selected via evolution for the very specific reason.
- The stability of the folded structure results primarily from the hydrophobic regions folding in together.
- Two proteins with different sequences can have the same sequence.
Conformations and Folding
- Protein conformational changes does not require breaking bonds.
- The peptide bond has partial double bond character and therefore are sterically hindered.
- Protein folding induces conformational changes in the backbone. The backbone torsion angles $\phi$ and $\psi$ determine that conformation of the protein chain.
- The Ramachandran diagram define the restrictions on backbone conformation.
- $\psi$ : TODO
- $\phi$ : TODO
- L: Left handed helix
- You see a lot of Glycines in loops as they are flexible because of the absence of any side chains.
- Prolines are in Cis conformation so they ususally “kink” the alpha-helix. Not generally found anywhere else.
- Secondary structural elements are conected to form simple motifs. Given motifs, you can create many folds.
- The interactions between the atoms in a protein control the folding of the protein into a well defined structure (native structure): Thermodynamic hyposthesis of protein folding. It was demonstrated by the famous Anfinsen Experiment which opened the folded structure of a protein and allowed it to relax in a controlled manner to recover the folded structure,
- This information is encoded in their amino acid sequence.
Evolution and Protein Structure
Reading Suggestion: The Molecules of Life (Chapter 5)
Protein structures are conserved during evolution while amino acids vary.
Motifs that are crutial for protein folding are conserved during evolution.
You can use protein globins to classify species at the molecular level.
Due to different selection pressures amd line seperation, protein sequences diverge between species.
How to quantify differences in sequences?
Use a substitution matrix (BLOSUM). We use a matrix of probailities. Each entry in the matrix is called a substitution score ($S_{ij}$). The higher the score (positive), the more likely is the substition.
The BLOSUM substitution score is the logarithm of the likelihood that one aa in a protein is replaced by another during evolution.
$S_{ij}$ is related to the frequency of substititon over random chance.
BLAST (Basic Local Alignmnet Search Tool) : Identify novel proteins, find functional relationships and sequence motifs and patterns.
TOP-7 is the first synthetic motif.
The most used measure is the Root Mean Square Deviation (RMSD).
Domains are “self contained units” that can be placed in different proteins.
PyMol Excercises
Superimposition is done by fitting the polypeptide backbone of your target protein on the reference protein. Best fit will have smallest Rms (root mean square) value.
What is PSIPRED?
Protein crystals have a high solvent content, which you can also observe as large spaces between the proteins in the Electron density maps.
Day 2
X-ray Crystallography
The further out spots the higher the resolution of the protein structure.
The dark ring ~3Å is caused by water molecules un the crystal.
- flowchart LR crystals --"x-rays"--> diffraction-pattern --> electron-density-maps-->atomic-structure atomic-structure --refinement-->diffraction-pattern
Methods to check crystallization conditions for proteins: Hanging Drop- and Sitting Drop- Vapour Diffusion.
Typical concentration for X-ray diffraction: TODO
High B-factor: low confidence in position of atom, which also corresponds to low electron densities.
In 2020 the number of structures in PDB reached 171,916.
Nuclear Magnetic Resonance (NMR)
Resonance Frequency in NMR: $$ R_f = \frac{2 \mu_BB_o}{h} $$
Resonance frequency depends on nature of the nucleus and its chemical environment.
Resonance frequency is directly proportional to the strength of the external applied magnetic field.
We commonly use $H^1$ and $C^{13}$ for labelling as they are NMR active.
Nuclear Overhauser Effect (NOE): Provides information on spatial proximity of protons in protein. Protons that are close by influence each others chemical shifts.
Crystallography si also applicable to very big proteins and complexes.
NMR: Dynamic ensemble of several energy-minimized structures, whereas, X-ray crystallography gives a static picture of protein.
NMR has a size limit: >25kDa is very difficult.
Cryo-EM
Cryo-EM prevents electron degradation of proteins.
Abbe’s Relationship: $d_R = 1.22 \lambda /NA$. Working out this formula gives us a $d_R$ to be 1.4Å.
flowchart LR biochemical-preperation --> cryo-em-sample-preparation --> Imaging --> data-collection(data-collection) dc(data-collection)-->image-processing-->reconstruction--> structural-analysis--> ModelGenerally Liquid Ethane is used.
3D images are reconstructed from 2D segment images from all angles.
Cryo-EM can do very big protein structures.
Bioinformatics and protein structure
Uniprot: Entries are manually annotated. This is the first stop to check an unknown protein. It contains references to other databases as well.- There are multiple
.pdbfiles for a single protein corresponsiong to all the different experiments done with the protein. They might have different resolutions, and precision. PhosphoSite Plus: provides comprehensive information and tools for the study of protein post-translational modifications (PTMs) including phosphorylation, acetylation, and more.STRING: All about protein interations. It contains protein-protein interaction data.PDB: The Protein Data Bank.Drugbank: Database of drugs.ChEMBL: Drug database where searches can also be compound-centric using different formats.
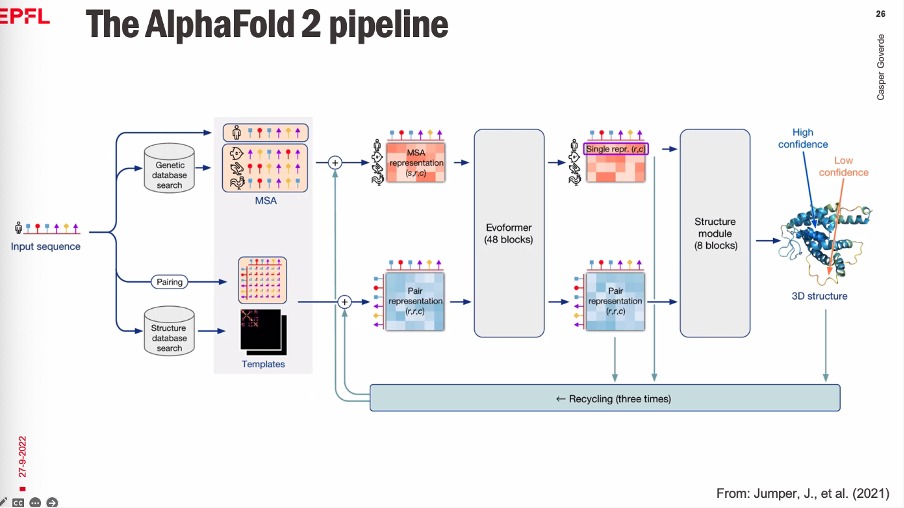
AlphaFold
Each step involves shrinking of the total phase space of that particular function.
- Critical Assessment of protein Structure Prediction (CASP) - competition held for prediction of Protein Structure. The FOM is GDT (Global Distance Test).
- AlphaFold 1 was the first to combine Multiple Sequence Alignment (MSA) and Deep Neural Networks.
- MSA finds similar sequence across different species, aligns, and then find “suitable mutations” or the set of all possible mutations (coevolution).
- If something mutates to a more positive base, the other base can mutate to something more negative, but not positive.
- We calculate distance matrix -> Correlation matrix.
- A neural network can approximate any function, given that your neural network has arbitrary width and we have an infinite dataset.
- AlphaFold also uses a Convolutional neural networks to recognise patterns.
- Output is the distance matrix, and the distance matrix of the two dihedral angles $\phi$ and $\psi$.
- The Input for AlphaFold-2 is the MSA.
AlphaFold-1:
AlphaFold-2:
Bad templates are inserted so AF won’t copy template features.
AMBER relaxation optimizes the final structure.
“Self-distillation training” - Using your own predictions as a dataset.
Human vs Other Species -> Prediction Score Comparision
Is all AlphaFold code open source? : Everything is opensource.
Autocompletion is based on the Attention Matrix, whcih comes from a language model.
AF also does a bunch of other things: protein-protein interactions, docking, predicting binding sites, etc.
ColabFold is a better version of AlphaFold 2. It is a lot faster with improved MSA step. It also allows for complex predictions.
Day 3
Binding
Spontaneous process at constant temperature and pressure always involves decrease in the Gibbs Free energy of the system ($dG < 0$).
It accounts for both enthalpic and entropic contributions of the system. $$ dG = dH - TdS $$
Equilibrium Constants:
$$ \Delta G^0 = -RTlnK_{eq} \rightarrow K_{eq} = e^{- \frac {\Delta G^0}{RT}} $$
- Homo Sapiens: ~15, 000 proteins expresses. 135,000 pp-interactions.
- Specificity is the ratio of binding affinities. They are theoreticlly not related. Innature, Higher affinity implies lower specificity.
- Binding sites usually have a hydrophobic core and then on the rim, you have bases that have specificity.
- Whwn fraction $f$ is half in a reaction, out dissociation constant $K_D$ equals the ligand concentration $L$.
- Univeral binding isotherm is expressed in terms of $K_D$.
Drug Binding by Proteins
- Find a Lead compound. That bind to the receptor. They are usually not very specific and affinative.
- Iterate the lead compound to imrove specificity and affinity.
- Competative inhibitors: $IC_{50}$ is the concentration of the inhibitor that reduces the binding of the protein by half maximal value.
- $IC_{50}s$ are used rather than a single value because the activity of the drug may vary between the population just because of random mutations.
- Energetics of the Binding: There is entropic loss but enthalpic gain. $\Delta G = \Delta H - T \Delta S$
- The affinity for a protein for a ligand is characterized by the dissociation constant, $K_D$.
- The strength of binding interactions is often correlated with hydrophobicity.
- MASIF: Surface-Based Protein-Protein Interaction Prediction
- All informtion required for predicting binding is present on the surface.
Exploring the potential of deep learning in de novo protein design
Protein perform a variety of functions
- Signalling
- Immunity
- Structure
- Catalysis
- Transport
Other functions (adapted for use):
- Therapeutics
- Biosensors
- Enzymes
- Biomaterials
Proteins as domains:
graph LR Sequence --> Structure --> FunctionAs a protein enginner you reverse the graph in your approach.
Motif grafting: Take parts of proteins (motifs) and combine them at the sequence level.
Classic de novo protein design is limited by already known structures (the PDB database). The top-down design method
A better approach is the de novo protein design: It takes a Bottom-up functional protein design. It is much more computationally expensive and suffers from a very low success rate.
Added an MC Step to the protein iteration step for scoring random mutations.
“Oracle models”
Exercise Notes
“MaSIF receives a protein surface as an input and outputs a predicted score for the surface on the likelihood of being involved in a PPI.”
— Assignment sheet
MaSIF can be used for iteratively for negative designing i.e. to remove features from the protein structure.
Day 4
DNA structure and replication
- The famous Photo 51 which was an X-ray crystallographic image of the DNA double helix strand.
- DNA replication proceeds only in one direction. It is because of the presence of phosphtes on the one side only, which is required for catalysis.
- The replication movement creates a “replication fork”, which leads to a leading strand (continuous synthesis) and lagging strand (discontinuous sequence). The fragments on the lagging strand are vcalled Okazaki fragments.
- Replisome;
- Average human chromosome: 150 Mbp
- Average Eukaryotic replisome speed: ~50 nt/s
- It will take 400 hours for the replication to occur.
- Hence there are 100s of origins for replication on a chromosome.
- How to identify DNA replication origins? (which are parts ) :
- Chop DNA, make some kind of loops with URA3. Only segments with Origins replicate and survive to grow into colonies.
- Deep Sequencing: sequencing of DNA at different time points gives rise to a frequency distribution, where the modes give you the Origin points.
- This method can also give you the speed of the fork , etc.
- With Cloning we can find all potential Origin sites, however, with Deep Sequencing, you find the Origins that are being actually used by the cell.
- End replication problem: Each time a cell divides, the chromosomes should be shortened. Finally the chromosomes should and be damaged.
- The telomere: Its a end point sequence that protects linear DNA from degradation by replacing the shortened DNA bases.
- Telomerase: the enzyme that does the replacement.
- Some cancers can express Telomerase to bypass the cell division limit.
- Telomers are repeated sequences. The T-loop hypothesis states that the end of the DNA strand loops back to itself to make a loop, so as to not signal as a broken DNA strand. The end-looping region is the Trlomer region.
- Different organisms also use different mechanisms to deal with end poins of its genetic material.
Day 5
Centromeres: from epigenetic to genetic
Cell division has been studied for a long time because of its importance in lifes’ processes.
Centrosomes and centromere are the two key factors for cell division.
The centromere is a protein/DNA structure which links chromosomes to spindle microtubules.
Centromeric architechtures are diverse:
- Regional centromeres
- Point centromeres
- Holocentromers
Kineticore: Protein complex TODO
If there are problems with Kineticores, you get aneuploid. ~90% of solid human tumors and ~75% of hematopoietic cancers are aneuploid.
Key steps for a centromere:
Centromere Position (decision on the location of the centromere, which is permanent)
Centromere function
Centromere integrity
The 171-bp alpha satellite is the fundamental unit of the centromere DNA.
~3% of the total human genome is chromomeric DNA. CENP-B is the only centromeric DNA-seq specific binding protein.
The centromeres exist in two domains: CENP-A and CENP-B.
Bigger centromere regions can lead to tangling of microtubles and chromatin during division.
Histones:
Histone mechanism" loading="lazy" src="https://www.genome.gov/sites/default/files/media/images/2022-05/Histone.jpg">
CENP-A and CENP-B arer particular histones that are specific to centromeres.
Where do Chromosomes go in interphase?
Cell Cycles" loading="lazy" src="https://cdn.kastatic.org/ka-perseus-images/6afac0fce62ec8a30c86f3e02c457224339bb697.png">
Interphase: Period between the cell divisions. During this phase, the genetic material (DNA) is replicated by the cell.
In mammals, you can find chromosomes that like to localize in a certain region of the nucleus.
How to build a mitotic chromosome?
mitosis means “threads”. The threads are the chromosomes.
Why cells need to build a mitotic chromosome?
- spatial compaction
- mechanical properties
- resolution of sister-chromatids
- switch off transcription
Interphase to mitosis compaction: 2X compaction.
Assembly of mitotic chromosomes is not fully known. There are several models:
- Hierarchical folding
- Scaffold/radial-loop
- Chromatin network
- Thin Layer stack
The localization of chromatin position explains how features far away in the genetic sequence regulate each other.
Positive supercoils are formed when the Replication Fork travels through the chromatin strands. To release these tension:
- Rotation of the replication fork itself.
- Nick the strand to release the tension and then re-attach.
- Topoisomerase II is required for decatenation and sister chromatin resolution
Transcription is turned off during Mitosis. Dogmas:
- Chromatin accessibility is limited.
- Global transcription suppression.